| RIKEN Center for Developmental Biology (CDB) 2-2-3 Minatojima minamimachi, Chuo-ku, Kobe 650-0047, Japan |
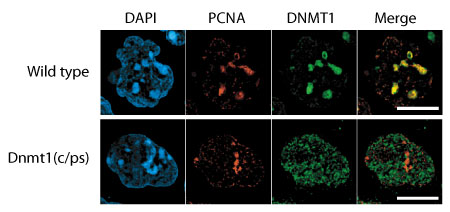
Recent work by Shin-ichiro Takebayashi in the Laboratory for Mammalian Epigenetic Studies (Masaki Okano; Team Leader). now brings an additional level of detail to our understanding of just how the DNMT1 enzyme achieves its developmentally-important maintenance function. His work, published in Molecular and Cellular Biology, reveals an essential role for the DNMT1 catalytic domain in mouse development, as well as the requirement for preexisting DNA methylation for the localization of DNMT1 to replicating DNA. Takebayashi began exploring the possibilities by making a subtle, single amino acid substitution in the catalytic center of the protein encoded by the DNMT1 gene, and used this to generate mutant embryonic stem cells and mouse embryos. He confirmed that in both the cells and the embryos DNA methylation levels were greatly reduced, similar to the case in DNMT1 null mutants. Even more tellingly, the catalytic domain mutant embryos were arrested shortly after gastrulation, failing to form somites and to undergo closure of the neural tube, phenotypes reminiscent of the null mutation. These findings point to a pivotal role for the catalytic domain in enabling embryonic development. The team examined the expression of multiple genes in their mutant mice and discovered a range of defects in the regulation of multiple genes, including imprinted genes and retrotransposons, as well as cell growth arrest. Looking at the localization of heterochromatin marks, they found that these too were globally affected; heterochromatin recruitment to pericentromeric regions was lower in all somatic cells. They also saw a small percentage of cells that exhibited hyperacetylation (a marker of transcriptionally active chromatin), a phenomenon that was never observed in wildtype cells. The picture that emerged was one of chromatin structural and gene regulatory abnormalities on the loss of DNMT1 catalytic function, leading to early embryonic lethality. Curious about how their mutant allele might affect DNMT1 recruitment to replication foci (where it is found in wildtype), they used immunofluorescence to track the mutated DNMT1 enzyme and discovered that it was instead diffusely present throughout the nucleus, suggesting that this defect was specifically attributable to the loss of the catalytic domain, rather than to some overarching functional change to the protein’s structure. |
|||||
|
|||||
 |
| Copyright (C) CENTER FOR DEVELOPMENTAL BIOLOGY All rights reserved. |