| RIKEN Center for Developmental Biology (CDB) 2-2-3 Minatojima minamimachi, Chuo-ku, Kobe 650-0047, Japan |
September 24, 2008 The advent of high-throughput genomics about a decade ago has yielded an extraordinary wealth of information on genome sequences, cDNAs, transcriptional start sites and binding sites for a growing spectrum of species. This burgeoning trove has simultaneously created the demand for new analytical tools, using computer algorithms to sift through the data, sometimes across genomes, in search of patterns of interest, identify new genes and regulatory factors and potentially synthesize new ones. As with any attempt to make sense of dizzyingly complex systems, such tools tend to work best when set to work on problems with at least some well-defined parameters. But once this combination of high-quality data sets and appropriate tools is in place, they represent a powerful new means of interrogating the molecular codes that underlie the machineries of life. Using the mammalian circadian clock as a model system, Yuichi Kumaki and Maki Ukai-Tadenuma of the Laboratory for Systems Biology (Hiroki R. Ueda; Team Leader) have done just that. In a study conducted in collaboration with scientists at the University of Pennsylvania, Kinki University and INTEC Systems Institute, the Ueda team constructed a database of DNA regulatory regions in mammalian genomes, surveyed it with statistical methods to predict new targets, and finally validated their model with synthetic regulatory elements. Their work, published in the Proceedings of the National Academy of Sciences USA, demonstrates the power of the systems biology approach in uncovering new biological principles.
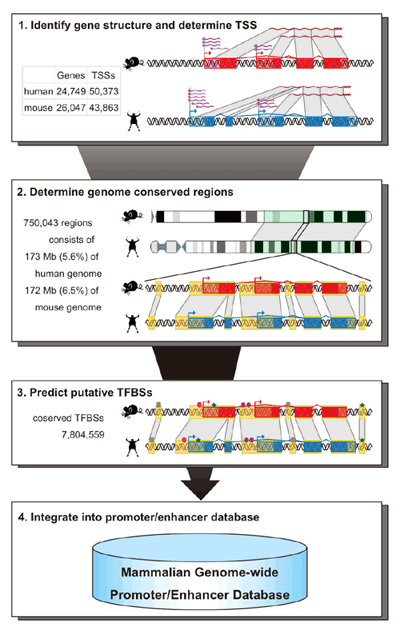
The oscillatory nature of circadian gene expression in mammals is maintained by the cyclical interaction of transcription factors, which produce periodic transcriptional read-out through three sets of elements whose activity peaks in morning (E-box), daytime (D-box) and night (RRE). While dozens of such clock-control elements are known, as an estimated 5-10% of mammalian genes show circadian expression, much remains to be learned. To address this question, the Ueda team assembled a database of human and mouse enhancers and promoters (two sorts of DNA regulatory regions), and customized a statistical method known as a Hidden Markov Model (HMM) to compare data and search probabilistically for new clock-controlled elements. They found that the Hidden Markov Model performed best in non-coding regions conserved between mouse and human, and that while D-box and RRE elements showed no bias in their distribution, E-box sequences tended to be grouped around transcriptional start sites. Their search turned up more than 6,500 predicted E-box, D-box and RRE elements. After winnowing out false positives, tests for clock-like temporal expression patterns using the top 100 putative clock-controlled elements for each of the three categories showed highly consistent peak expression times in approximate phase with other circadian elements. The team next validated their candidates in vitro, picking the ten highest-confidence E-box, D-box, and RRE elements, and transfecting them fused to a luminescent reporter into cells to observe their transcription. After stimulation to synchronize the cells circadian rhythms, 4 of the transfected E-box candidates, 7 of the D-box, and 6 RREs showed strong daily oscillations. They next checked the expression of these 17 genes in vivo, examining endogenous transcripts from seven different mouse tissues and found circadian patterns of expression for 13. As systems biology seeks not only to find what is in a system, but also conceptually what might be, the Ueda team looked back to the HMM. The Hidden Markov Model is essentially a means of inferring probabilities about unknown states in a system by looking at known properties of the same system, such as probabilities of the state generating a given outcome (emission) and of transition from state to state. Kumaki and Ukai-Tadenuma emitted sequences from the E-box, D-box and RRE models, and filtered out the naturally occurring ones, leaving only those that appeared not to be present in the genome (but which nonetheless are predicted to have similar activity to their natural counterparts). Taking one high-scoring and one low-scoring element for each of the models, both of which contained the same consensus sequence but different flanking sequences, they tested them in a synthetic reporter system and found that all of the high-scoring elements showed high-amplitude circadian transcriptional activity. This synthetic reporter assay clearly provided the evidence that the flanking sequences around the consensus sequence can function to titer the amplitude and oscillating rhythm of their transcriptional output. Thinking that such flanking sequences might play a role in changing the binding affinity (and thereby alter the amplitude) of clock gene regulators, the team analyzed the affinities of activators and repressors in competitive binding assays. Interestingly, among the E-box elements (but no D-box or RRE), those which had previously been classed as low-scoring had higher affinity for activators and approximately normal affinity for repressors, while the affinities for activators and repressors in high-scoring E-box elements was closer, suggesting that a balance between direct activators and repressors is important for generating high-amplitude E-box output.
|
||||||
|
||||||
 |
| Copyright (C) CENTER FOR DEVELOPMENTAL BIOLOGY All rights reserved. |