| RIKEN Center for Developmental Biology (CDB) 2-2-3 Minatojima minamimachi, Chuo-ku, Kobe 650-0047, Japan |
October 25, 2008 For proteins and other biological molecules, function follows on form. Scientists have simulated the molecular dynamics by calculating interactions between atoms and determining their stability in different conformations and the likelihood of transitions between states. One method for analyzing simulated data, known as principal component analysis (PCA), which uses the individual atoms positions (atomic coordinates) works well at extracting conformational fluctuation and predicting changes in response to perturbations, but runs into difficulties when dealing with major transitions, such as those involving multiple stable conformations. Yohei M. Koyama of the Laboratory for Systems Biology (Hiroki R. Ueda; Team Leader) has now introduced a version of PCA that takes advantage of the potential energy in atom-atom interactions, an approach he has named PEPCA (for potential energy PCA). The work, which allows for the analysis and prediction of major conformational changes, is published in Physical Review E.
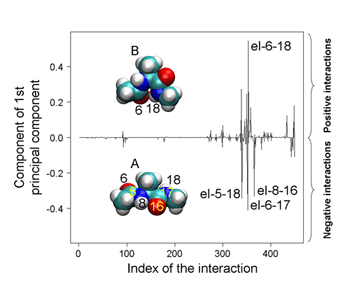
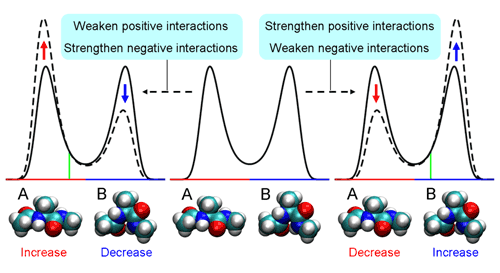
PCA using atomic Cartesian coordinates works well in analyzing conformational fluctuations around a single stable conformation. But since these coordinates depend on the overall molecular motion, the confounding factors of translational and rotational motion must first be eliminated. And while this is possible even in large molecules for translational motion, factoring out rotational motion has proven to be a knottier problem. Koyamas approach was first to look at the traditional method and make some generalizations, in which he determined that PCA can be used as a means of finding perturbations that cause the largest conformational change. In order to avoid the problems with conventional PCA, he developed an alternate to atomic coordinates, using the potential energy of interactions between atoms as a surrogate. And, as this energy is determined solely by the relative positions of a pair of interacting atoms, it is unaffected by overall molecular motion, meaning that the problematic rotational motion does not come into play. To show the utility of PEPCA method, Koyama focused on a conformational transition in alanine dipeptide as his first test bed. He discovered in his analysis that the perturbation that induced the largest conformational change involved a change in the distribution of a pair of stable states, and identified important interactions in determining the stability of and transitions between these two conformations, demonstrating the value of the new approach in both identifying and predicting molecular conformational change.
With the development of high-speed computational facilities specializing in molecular dynamics simulations, were now increasingly able to simulate physiologically significant conformational changes, says Ueda. And I think that the PEPCA method will help in determining the atomic interactions that are critical to molecular function, which could be of value, for example, in rational drug design. |
|||||||||
|
|||||||||
 |
| Copyright (C) CENTER FOR DEVELOPMENTAL BIOLOGY All rights reserved. |